HELP GUIDE
The PRDB is a regularly updated database of all the protein repeats found by the T-REKS program in large databanks.This webpage contains many options and features to browse the protein tandem repeats of the database. It includes selective criteria, a query submission and a display of results based on AJAX technologies, and a toolbox for user-defined queries.
1- The form and the options:
These options are for both quantitative and qualitative criteria, and can be divided into three categories: (1) general options, (2) options related to the tandem repeat itself (TR),(3) options related to tandem repeat-containing protein (RCP).
All selected/defined options are combined by the logical operator "AND", which means that some of the combinations can lead to unsuccessful results.
The default values are meant to display all the repeats from the SwissProt database.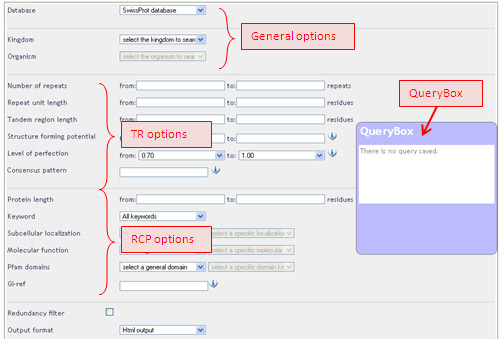
General options:
- Databank option: allows the user to choose a protein sequence databank explored by T-REKS Jorda J, Kajava AV, October 2009. T-reks: identification of tandem repeats in sequences with a k-means based algorithm. Bioinformatics 25 (20), 2632-2638 .
- NR: Non-redundant databank from NCBI which is one of the most complete and is constituted by nonredundant Genbank CDS translations, SwissProt and PDB.
- Swissprot: A highly-annotated, curated protein sequence database available at Expasy website. It features a minimal redundancy and a high level of integration with other databases.
- PDB: Protein sequences from the Protein DataBank PDB, a database of experimentally determined three-dimensional structures of biological macromolecules. More information at the PDB website.
- Kingdom option: allows the user to select one or several kingdoms. It integrates major subkingdoms like Fungi, Metazoa, Plants, Glaucocystophyceae, Rhodophyta , Protozoa and Viridiplantae.
- Organism option: Is enabled once a kingdom is selected. It only displays the organisms related to the selected kingdom.
TR options:
These options are linked to criteria specific to the protein TR, or a range of values in the case of quantitative criteria. For the latter, exclusive lower and upper bounds can be set to allow only values in between.
- Number of repeats: The number of repeat units in the TR.
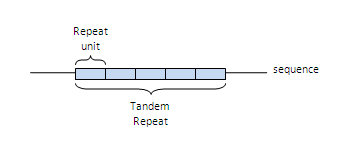
- Repeat unit length: The average length of the repeat units in the TR.
- Tandem region length: The length of the whole TR, it usually corresponds to the number of repeats * repeat length.
- Structure forming potential: this value is based on the TOP-IDP scale defined by Campen et al (Campen A, Williams RM, Brown CJ, Uversky VN, Dunker AK: TOP-IDP-Scale: A new amino acid scale measuring propensity for intrinsic disorder.Protein and Peptide Letters 2008, 15(9):956-963). TOP-IDP measures the propensity of a sequence to be intrinsically disordered. The Structure forming potential is the TOP-IDP value of a TR region.
- Level of Perfection: TRs are rarely perfect due to a number of mutations. The level of perfection to which they are repeated is determined by using a similarity coefficient called PsimJorda J, Kajava AV, October 2009. T-reks: identification of tandem repeats in sequences with a k-means based algorithm. Bioinformatics 25 (20), 2632-2638. This coefficient, based on Hamming Distance, varies from 0 to 1(1 is the value for perfect repeats). However, the coefficient values start from 0.7 in PRDB, because below this point, the number of false positive TR becomes significant.
- Consensus pattern: The consensus sequence of the TR. It is derived from the multiple alignment of the repeat units. Each residue of the consensus is determined by picking the most frequent residue from its corresponding column in the alignment. (The indel character '-' is considered as residue). In ambiguous case, the residue will be noted X.
- Redundancy filtering: Sequenced strains of the same organism can lead to the presence of redundant sequences in PRDB. Subsequently, certain types of sequences can be overrepresented. This option allows the user to ignore the duplicates in the results.
RCP options:
- Protein length: the RCP sequence size.
- Gi-ref: The sequence identifier of the RCP provided by NCBI
- Subcellular localization: the compartment where resides the RCP in the cell. This information comes from the Genpept file.
- Molecular function: Activities that can be performed by a RCP. Genpept files of Swissprot proteins often contain GO accession number. These GO numbers linked to the Gene ontology database give us a clue of what are the molecular functions of a protein (see details). The families have been selected from the ontologies in which the molecular functions are involved.
- Pfam domains: the PFAM domains found in RCPs (Finn, R. D. et al. The pfam protein families database. Nucl. Acids Res. 38, D211-222 (2010)) by hmmpfam. This program is part of the HMMer package, and has been used to launch the RCP sequences against the HMM database to identify their PFAM domains. These values have been organized by text classification (based on frequency counts of the words of the pfam names) and by using the pfamclans.
- Keyword: Keywords provided by authors in the Genpept file.
Next options are only available for the SwissProt databank. Most of them are related to data that has been retrieved from the parsing of Genpept XML files hosted in the NCBI server. This data is classified in families in order to simplify the queries and give a hierarchical view.
Output options:
- Html: Default output. It displays the TRs as a list in a HTML webpage. This webpage is called via an AJAX request allowing the user to keep his query in the form which could be lost by a page refresh.
- Csv: outputs all TRs in a csv file. The results can be exported with user-defined fields. The first record is the header containing the field names while the following lines are the TR values separated by a 'TAB'.
- Fasta: outputs a FASTA sequence file of the repeated regions or the RCP sequences.
2- The QueryBox:
This tool is always accessible to the right of the form. It was developed to manage the different TR queries that have been saved in the session (this option is available on the results page).Selected queries can be deleted or combined through three actions:
- Intersect: returns a set of TRs common to all the queries.
- Exclude: returns the symmetric difference of TRs issued from selected queries.
- Merge: returns the TRs of all queries in one set.
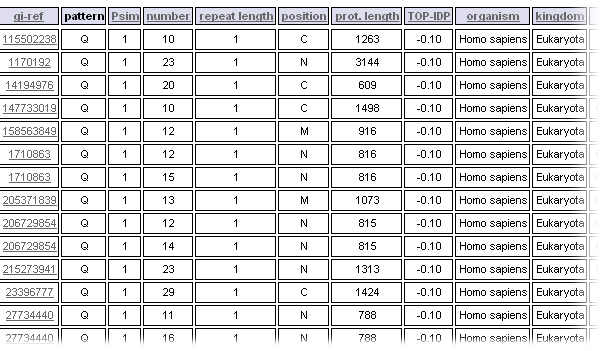
3- The results list:
Results of the query are listed in a table, the data can be sorted by clicking on a field in the header.
- The position criterion gives information about the localization of the TR in the protein sequence. 'N' is for N-terminal third, 'M' for the middle third and C for C-terminal third of the considered protein. If the TR is located in two or all regions, the position will be correspondingly a combination of 2 or 3 characters.
Polyglutamine repeats found in cytoplasmic proteins in Humans
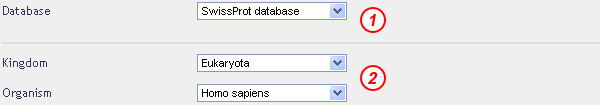
Select the databank to explore, "Eukaryota" in the kingdom field and the "Homo sapiens" organism
Set the upper limit for the repeat unit length to 1, polyglutamine repeats being runs of a single glutamine residue.

Set the consensus pattern to Q

Get only the proteins localized in the cytoplasm

Set the output to html - Display the results